
 分享
分享

患者郑某某,女性,48岁,农民。2019年01月16日主因「咳嗽、咳痰伴胸闷、气短20余日」入院。
现病史
患者20余日前因劳累受凉后出现咳嗽、咳痰,为白色泡沫样痰,痰液粘稠不易咳出,量中,无痰中带血及咯血。伴发热,自测体温最高达38.5°C,无寒战、抽搐,无午后低热。伴活动后胸闷、气短,且进行性加重,无胸痛、心悸,无黑朦、晕厥等不适。于2018年12月24日就诊当地医院诊断「双肺多发结节性质待定 肺部感染」给予抗感染止咳化痰治疗1周余,患者体温恢复正常,但仍咳嗽、咳痰,复查胸部CT提示肺内病灶无改善。为求进一步诊治就诊我院。
既往史
患者贫血病史数年,曾诊断为「缺铁性贫血」,不规律口服「蔗糖铁」治疗(具体不详)。否认冠心病、糖尿病、高血压病史。否认药物、食物过敏史。否认手术史。
生育史
生1儿1女,爱人及子女均体健。
个人史
否认工业毒物、粉尘、放射性物质接触史。饲养猪20年余。否认吸烟、饮酒史。
家族史
母亲因血液系统疾病于1年前去世(具体不详),父亲健在。1个姐姐,2个弟弟均体健。
体格检查
体温36.5℃,脉搏86次/分,呼吸22次/分,血压125/70mmHg,SPO2 91%(未吸氧)。贫血貌,口唇无紫绀,双肺呼吸音粗,双肺可闻及散在湿啰音。颈部及双侧腋窝淋巴结未触及肿大。双侧腹股沟区均可触及约1×1cm肿大淋巴结,质中,无压痛,活动度尚可。心脏、腹部及神经系统查体未见异。无关节红肿。全身无皮疹。双下肢轻度凹陷性浮肿。
辅助检查
肺功能:常规通气功能及气道阻力正常;小气道功能早期改变;轻度弥散功能障碍,FVC% pre:100%,DLCO:76%,FEV1/FVC:81%,FEV1% pre:95%。
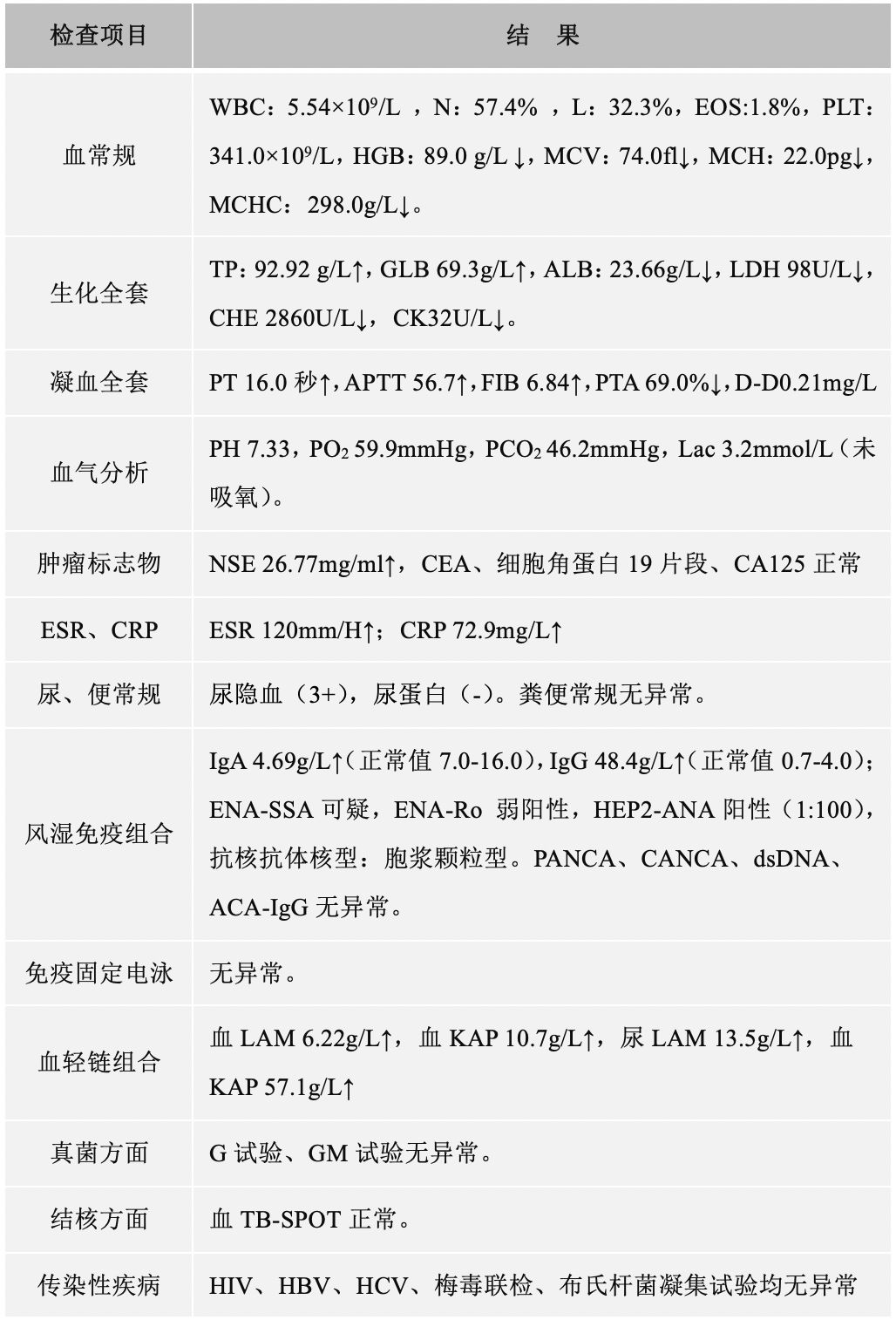
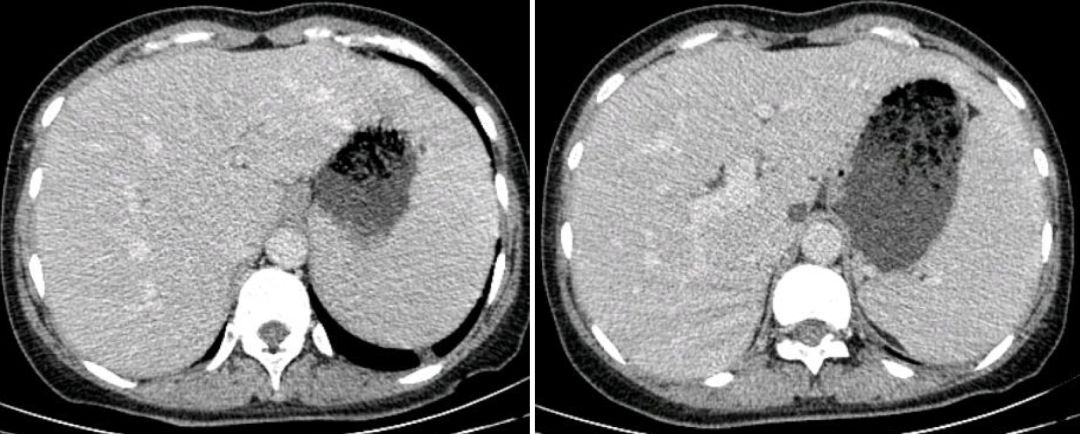
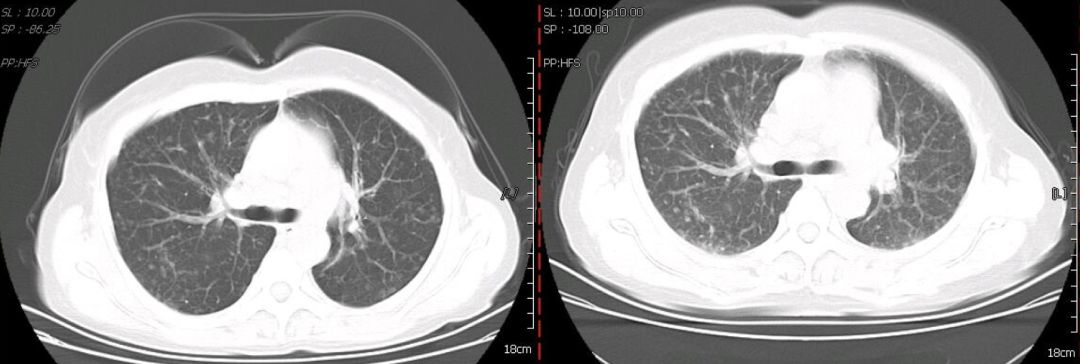
心脏彩超:心脏结构、运动及血流分布未见明显异常;左室舒张功能减低;左室收缩功能正常。腹部彩超:肝脾增大。妇科彩超:子宫肌瘤、宫颈囊肿。胸部CT(2019年1月4日):双肺野内多发大小不一,密度不均,小叶中央型及小叶周围型结节影。纵膈内多发肿大淋巴结,增强后见明显均匀强化,淋巴结未见明确融合改变。双侧腋窝多发肿大淋巴结。肝脏及脾脏增大(图1)。
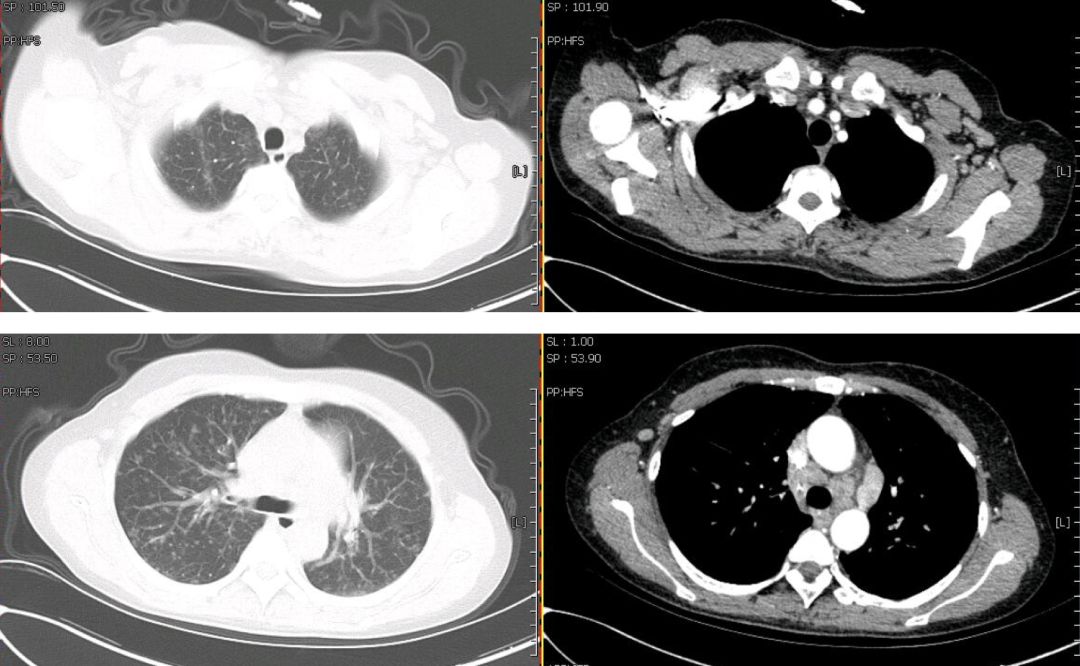
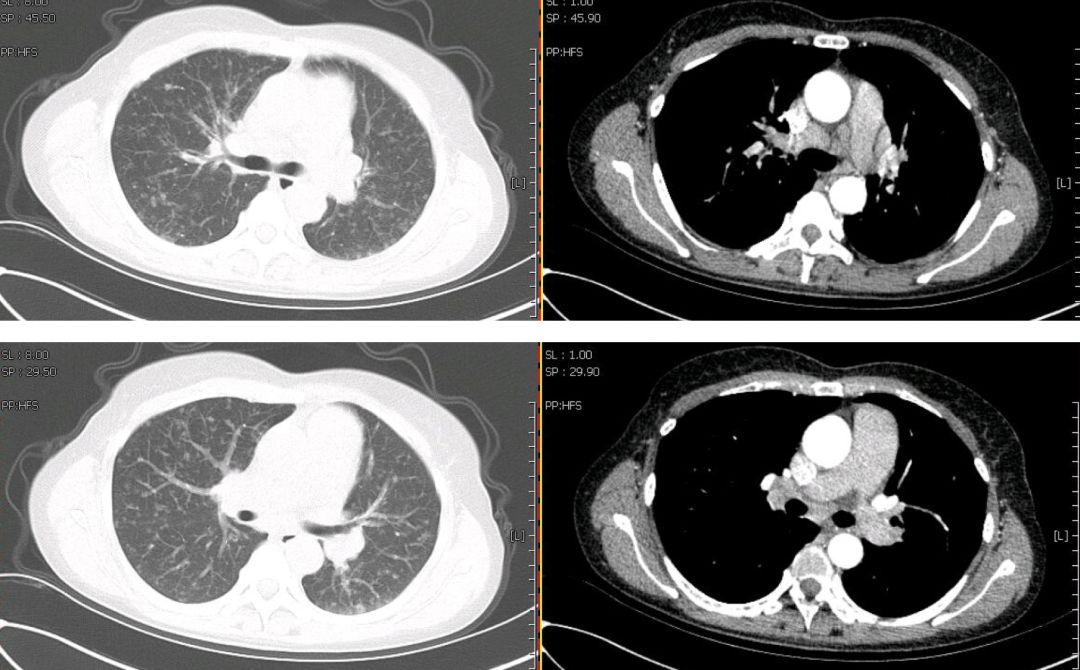
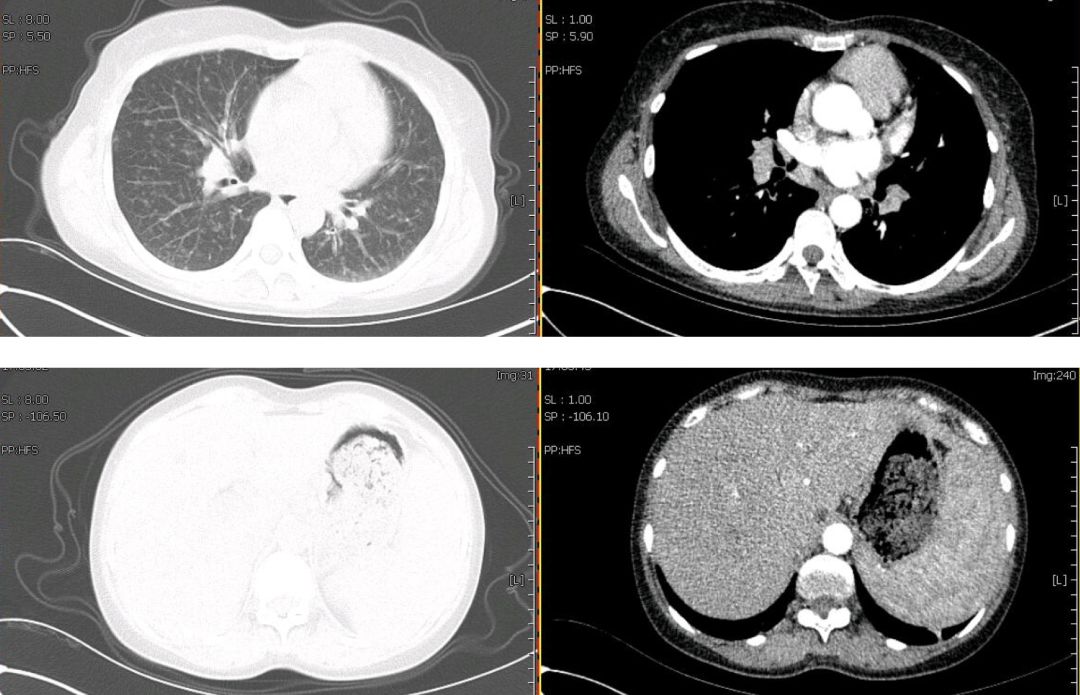
【图1】胸部增强CT(2019年1月4日):双肺多发结节,纵膈、腋窝多发肿大淋巴结
电子支气管镜检查:左右主支气管管腔可见大量脓性分泌物,右中叶支气管粘膜肥厚、充血,管腔通畅,右下叶背段支气管粘膜充血,管腔通畅(图2)。肺泡灌洗液细胞学:白色、微浊,白细胞总数3040个/mm³,中性粒细胞79%,巨噬细胞13%,淋巴细胞4%,嗜酸性粒细胞4%。肺泡灌洗液结核分枝杆菌PCR检测:未检出结核分枝杆菌。肺泡灌洗液培养:草链、奈瑟菌属。
【图2】电子支气管镜检查
进一步检查
1、EBUS-TBNA(图3)。
【图3】电子支气管镜下EBUS-TBNA
EBUS-TBNA病理:(4R淋巴结)纤维素、红细胞、淋巴细胞及淋巴组织,另见少量软骨组织。免疫组化:TTF-1(-),CD56(-),Syn(-),CgA(-),Ckpan(散在+),LCA(+),Ki67(index约10%)。特殊染色:PAS(-)(图4)。
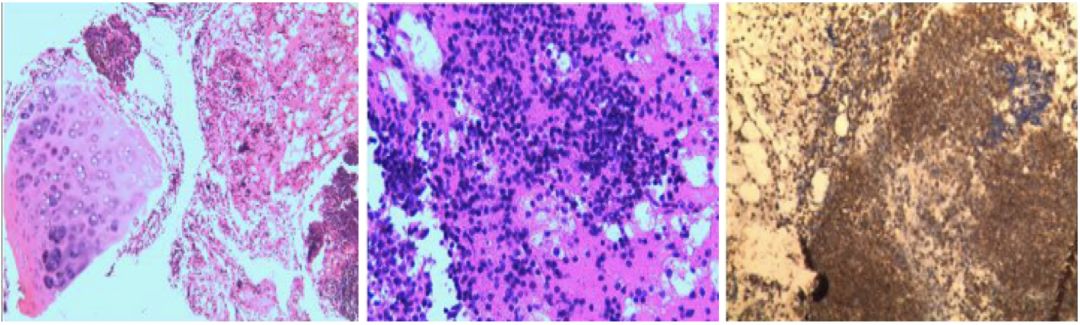
【图4】EBUS-TBNA(4R淋巴结病理结果)
2、骨髓穿刺+活检:粒红巨三系增生活跃,分类可见形态异常浆细胞9%。粒系增生活跃,各阶段比值及形态大致正常;红系增生活跃,以中晚幼红细胞为主;淋巴细胞占11%,形态正常;巨核细胞正常,血小板可见。骨髓染色体核型分析:未见克隆性异常(图5)。
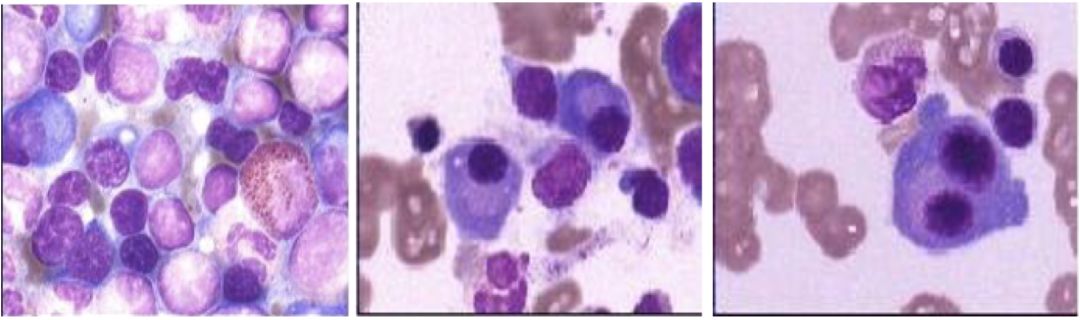
【图5】骨髓穿刺+活检结果
3、全身骨断层扫描:未见异常。
4、全腹增强CT:肝内多发囊肿;脾脏增大,腹主动脉旁、双侧髂血管旁及腹股沟区淋巴结肿大,均匀强化;左肾小囊肿;子宫肌瘤,宫颈那氏囊肿(图6)。

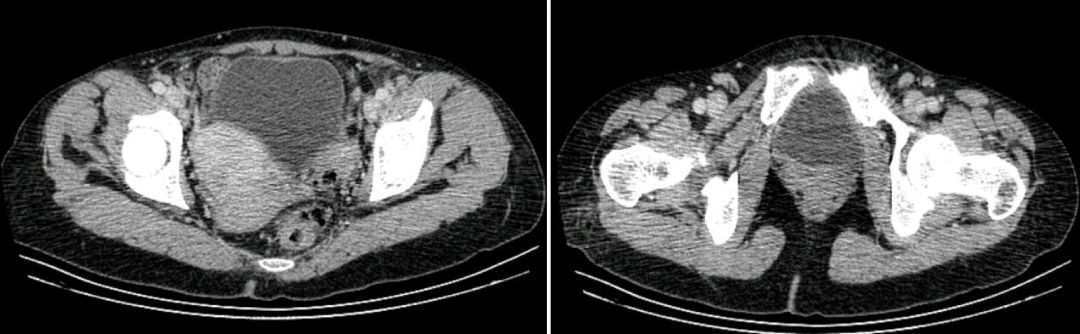
【图6】全腹增强CT扫描
5、右侧腹股沟淋巴结活检:淋巴结组织,淋巴滤泡增生,滤泡见可见大量浆细胞样细胞浸润,结合免疫组化检查结果,符合淋巴结反应性增生并大量浆细胞浸润。免疫组化:CD3(T淋巴细胞+),CD20(B淋巴细胞+网+),Ki67(淋巴滤泡生发中心+),Ckpan(-),Bcl-2(淋巴滤泡生发中心-),EBER(-),CD38(浆细胞+),CD138(浆细胞+),Lambda(+),Kappa(+),CD10(-),Bcl-6(淋巴滤泡生发中心+)(图7)。
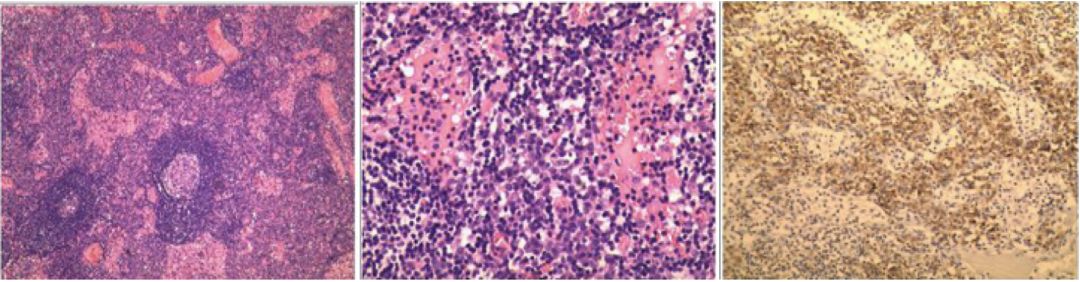
【图7】腹股沟淋巴结(右侧)活检病理结果
6、血清球蛋白Ig4亚类7.7g/L (正常范围0.02-2.0)。
诊断思路
多发淋巴结(纵膈、腹腔、腋窝、腹股沟淋巴结)肿大及多器官及系统累及(肺脏、血液系统、肾脏、脾脏),原因?
IgG4 相关性疾病?
淋巴瘤?
结节病?
结核?
其他?
1、IgG4相关性疾病?
IgG4相关疾病(IgG4-related disease,IgG4-RD)是一种自身免疫介导的炎性纤维化疾病,可以造成器官肿大、组织破坏甚至器官功能衰竭。该患者骨髓及淋巴结可见浆细胞细胞浸润,且血清球蛋白Ig4亚类明显升高,故需高度怀疑此病。但IgG4-RD是一种累及多器官和组织、慢性、进行性进展的自身免疫性综合征,已报道的相关系统表现包括硬化性涎腺炎、肺部炎性假瘤和间质性肺炎、自身免疫性胰腺炎、腹膜后纤维化、硬化性胆管炎、硬化性胆囊炎等。但该患者并无上述相关系统表现,且确诊需依靠病理特异性表现(即器官或组织中存在广泛的 IgG4 阳性浆细胞增生性浸润及硬化),故需进一步完善病理明确诊断。
IgG4-RD分类标准:
1、一或多个器官出现弥漫性/局限性肿胀或肿块的临床表现;
2、血清IgG4浓度≥1350mg/L;
3、组织病理学检查:①显著的淋巴细胞、浆细胞浸润和纤维化;②IgG4阳性浆细胞浸润:IgG4阳性/IgG阳性细胞>40%,且IgG4阳性浆细胞>10个/高倍镜。
本例患者符合1+2标准,疑诊IgG4-RD,但该患者并无上述相关疾病系统表现,IgG4阳性/IgG阳性细胞<40%,且确诊需依靠病理特异性表现(即器官或组织中存在广泛的 IgG4 阳性浆细胞增生性浸润及硬化,如果HE染色下看到席状纤维化、闭塞性脉管炎可以提高诊断的特异性),故需进一步完善病理明确诊断。
Castelman病(Castleman Disease,CD)是一种不明原因的淋巴组织增生性疾病,可累及单个或单组淋巴结而表现为局灶型病变,也可累及多中心淋巴结或淋巴结外器官而表现为多中心性病变。本例患者多发淋巴结肿大,且多系统受累,且合并免疫球蛋白血症、免疫学指标异常,因此需要鉴别此病。但CD最常累及纵隔及肺门淋巴结,表现为局灶型巨淋巴结增生,而发生于肺实质的CD非常罕见。本例患者表现为双肺多发实性小结节及磨玻璃结节性病变,并未见肺内较大实性结节或肿块,CD病疑似,但亦需进一步病理明确诊断。
2、淋巴瘤等恶性血液系统疾病?
淋巴瘤是起源于淋巴造血系统的恶性肿瘤。该患者存在无痛性淋巴结肿大,肝脾肿大,且全身多组织器官受累,因此,该病亦需高度怀疑。但淋巴瘤肺累及,主要表现为圆形或类圆形或分叶状阴影,病变进展可压迫支气管致肺不张,有时肿瘤中央坏死形成空洞,极少部分患者表现为弥漫性间质改变。该病确诊主要依靠骨髓活检及淋巴结病理检查。该患者骨髓涂片、活检、染色体核型分析以及淋巴结活检均未发现淋巴瘤细胞,但骨髓及淋巴结可见浆细胞浸润,需进一步完善淋巴瘤免疫分型等进一步排除诊断。
3、结节病?
结节病是一种非干酪性坏死性上皮细胞肉芽肿炎症性疾病,以侵犯肺实质为主,并累及全身多脏器。该患者多脏器受累,且存在贫血、ESR增快、血清球蛋白增高、血清白蛋白降低,但该患者血钙、血清碱性磷酸酶正常,肺泡灌洗液细胞分类以中性粒细胞为主,EBUS-TBNA以及腹股沟淋巴结活检均未查见非干酪性坏死性肉芽肿,因此结节病可基本排除。
4、结核病?
该患者病程中有发热,经过广谱抗感染治疗后虽然发热、咳嗽、咳痰症状有所缓解,但肺内呈多发的、分布不均匀的结节性病变,且纵膈淋巴结明显增大,ESR、CRP明显升高,需要考虑结核病。但患者无消瘦、盗汗症状,T-SPOT阴性,且淋巴结活检病理未发现干酪坏死性肉芽肿等结核特征性病变,因此目前可基本排除结核诊断。
诊断关键仍然是病理!如何进一步明确诊断?
请南京医科大学附属鼓楼医院病理会诊
会诊结果:(纵膈淋巴结EBUS穿刺活检):送检较多淋巴细胞,未见明确恶性依据。(右侧腹股沟淋巴结活检):送检淋巴结结构大致正常,副皮质区级髓质区大量浆细胞浸润,局灶皮质区见葱皮样淋巴结小结,少数生发中心见透明血管。免疫组化: κ(++),IgG(++),IgG4(+),IgG4:IgG约30%。结合临床、影像学、淋巴结活检及免疫组化结果,考虑浆细胞性Castleman病伴分泌性IgG4的浆细胞增多。
进一步治疗
因考虑患者为Castleman病,给予醋酸泼尼松50mg qd治疗,患者自觉胸闷、气短、咳嗽、咳痰症状明显缓解。1月后复查胸部CT(图8),与1月24日胸部CT相对比可见双肺结节呈增多趋势,且双肺间质纤维化明显。提示单用激素治疗无效。
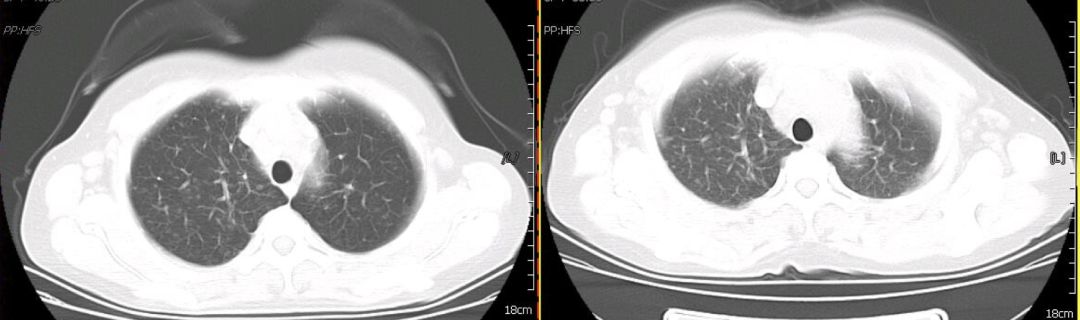
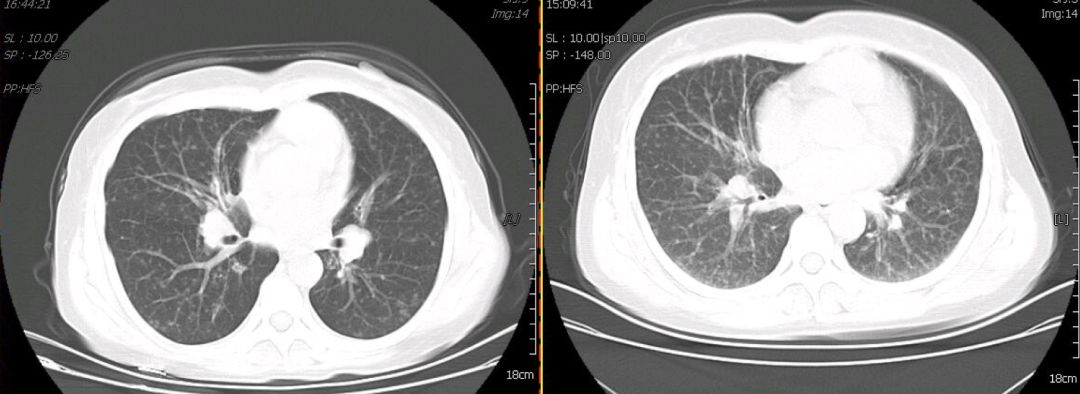
【图8】激素治疗后复查胸部CT(左侧为1月24日,右侧为3月6日)
确诊结果与疑问
病理确诊为:Castleman病
疑问:1、肺内多发小结节病灶是否是Castleman病肺部累及?2、激素治疗为何效果不佳?
进一步检查
与患者及家属沟通后行外科胸腔镜肺组织活检,病理结果:(右上肺)肺组织,部分区域肺泡间隔增宽,间质纤维组织轻度增生并多量淋巴细胞、浆细胞浸润,部分区域炭末颗粒沉积(图9)。
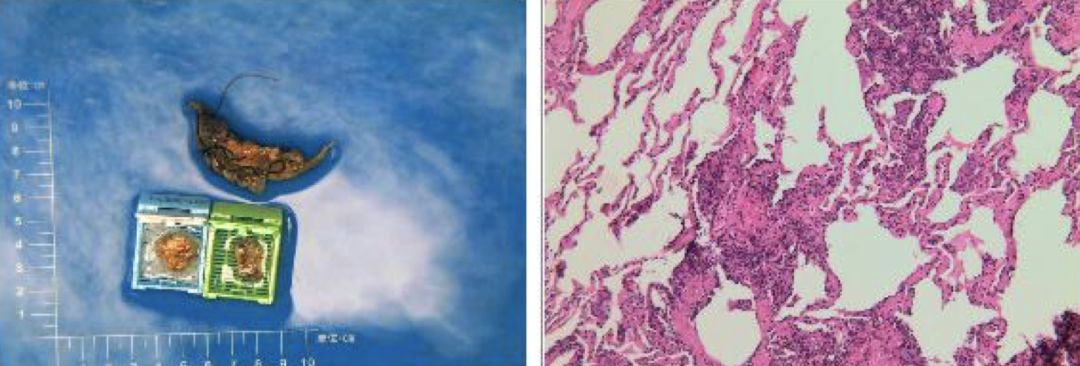
【图9】肺组织活检病理
明确诊断
Castleman病(多中心型,浆细胞型)
随诊:目前该患者正接受CHOP方案全身化疗。
病史回顾及文献复习
本例患者,中年女性,以咳嗽、咳痰、胸闷、气短为主要临床表现,完善相关检查后提示双肺多发磨玻璃结节,且伴随淋巴系(纵膈、腋窝、腹股沟、腹主动脉旁、右侧髂血管旁淋巴结)、骨髓、肝脏、脾脏、血液系统、肾脏系统累及。排除了结节病、结核病、转移瘤等常见疾病后,最终根据腹股沟淋巴结及肺组织活检病理明确诊断为Castleman病(Castleman Disease,CD)。
CD也称巨大淋巴结增生症或血管滤泡性淋巴结增生症,是一种罕见的病因及发病机制未完全明确的慢性淋巴组织增生性疾病。由Benjamin Castleman(1906~1982)于1954年首次报道 [1]。目前CD发病机制尚不完全清晰,但研究报道白细胞介素(IL-6)、人疱疹病毒(HHV)以及HIV感染在CD的发病中起重要作用 [2-4]。
临床上根据肿大淋巴结的分布范围,CD分为单中心型(Unicentric CD,UCD)和多中心型(multicentric CD,MCD)。
UCD常见于30岁~40岁患者,平均诊断年龄34岁(范围,2~84岁)。UCD具有轻微的女性优势(男女比为 1:1.4)。MCD是一种全身性疾病,通常出现于60岁左右,具有轻微的男性优势。约1/3 MCD患者发展为恶性肿瘤,如Kaposi肉瘤、恶性淋巴瘤等 [5, 6]。
根据组织病理学的特点,CD分为透明血管型(HV-CD)、浆细胞型(PC-CD)、混合型、浆母细胞型 [7]。
透明血管型占全部病例90%。浆细胞型仅占10%,多表现为多中心型,往往累及机体多发脏器。大多数的UCD病例在组织学上归类为HV-CD,HV-CD由于滤泡内和滤泡间的透明质化小血管数量的增加而引起的,并伴有髓窦的闭塞。淋巴滤泡数量增多,表现出「退化」的特征。套膜区小淋巴细胞在生发中心周围常呈同心圆排列(「洋葱皮」表现),滤泡可被血管呈放射状穿透(「棒棒糖」样滤泡)。滤泡间区可见浆细胞,但通常数量较少,呈小簇分布。具有丰富浆细胞的病例可能反映了在透明血管和浆细胞组织学变异之间具有「混合」或「过渡」特征的UCD [8]。
UCD主要表现为单部位淋巴结肿大,缺乏特异性临床表现及实验室指标异常,多为体检发现。MCD多表现为弥漫性淋巴结病,可伴随脾脏肿大,B类症状(发热、盗汗、体重减轻),POMES综合征(多发周围神经病、免疫球蛋白血症、脏器肿大、内分泌病变、皮肤损害),约15%~25%的MCD患者存在POMES综合征 [6, 9, 10],另外可见腹水和(或)胸腔积液,贫血,炎性指标升高,高丙种球蛋白血症,低蛋白血症,免疫学指标异常等。
特发性MCD的国际共识治疗指南 [11] 指出,满足下列5项标准中至少2项及2项以上则可诊断为重症MCD患者,否则诊断非重症MCD:
①ECOG评分≥2分;
②IV期肾功能不全(eGFR <30;肌酐> 3.0);
③全身水肿和/或腹水和/或胸膜/心包积液(高细胞因子血症/低白蛋白的影响);
④HGB≤8.0g/dL;
⑤肺受累/间质性肺炎/呼吸困难。
Xuanye Zhang [12] 等临床调查研究显示,年龄较大(≥40岁)、浆细胞或混合组织学类型,存在B型症状、脾肿大、腹水和/或胸腔积液、低白蛋白血症和高球蛋白血症的MCD患者预后较差。
根据诊疗指南,本例患者符合重症MCD,评估预后较差。
2015年NCCN非霍奇金淋巴瘤诊疗指南 [13] 指出,CD病诊断需满足以下三个方面:
1、对所有切片进行病理学检查(至少1个含淋巴增殖性疾病组织的石蜡块;如果认为所获标本不足以确诊,则需重新活组织检查)。
2、单独细针穿刺(FNA)或空芯针活组织检查不宜作为CD初次诊断的依据。但是在某些情况下,当淋巴结难以切除或切取活组织检查时,联合FNA或空芯针活组织检查并结合辅助检查(IHC、FCM、PCR、ISH等技术)可以为诊断提供充分的信息。
3、确诊必须依赖于充分的免疫分型。IHC抗原谱有:Kappa/ Lambda 、CD20、CD3、CD5、CD138、人疱疹病毒8潜伏期相关核抗原1(HHV-8 LANA-1)等。
本例患者诊疗初期曾行TBNA-EBUS,但病理未提示明显异常病变,后行腹股沟淋巴结活检才明确淋巴滤泡增生,且淋巴滤泡间大量浆细胞浸润,故由此可见对于疑诊断CD患者,有条件者需尽量行淋巴结切除或活检。
肺部CD以UCD最为常见,约占60~70%。最常见的部位是胸腔(63%),以中纵膈与肺门多见,其次是前纵膈和后纵膈。
CT扫描对该病的诊断和鉴别诊断有重大价值,主要表现为 [14-16]:
(1)单发较大的软组织肿块,边缘清楚;
(2)增强扫描瘤灶多呈显著均匀强化;强化程度可与同层面的动脉相同;很少侵犯周围组织;
(3)5%~10%病例瘤灶中央可见呈簇状分布的分支状钙化,即特征性的树枝状钙化;
(4)病灶内很少发生液化及坏死。
肺实质累及 [17]:发生于肺实质的CD罕见,仅有极少数的报道,多见于MCD [17, 18]。其CT扫描主要表现为磨玻璃样病灶(GGO)、边缘模糊并呈小叶中心性分布的小结节、支气管血管束增厚、小叶间隔增厚、气腔实变以及薄壁肺气囊。CD肺内侵犯的组织病理学改变为淋巴细胞性间质性肺炎(LIP),主要为大量淋巴细胞和浆细胞在肺实质内浸润。
本例患者为双肺肺实质累及,胸部CT主要表现为多发磨玻璃结节,大小不一,密度不均,分布无规律,且伴纵膈多发淋巴结肿大,与田欣伦 [19] 及赵爽 [20] 文献报道非常相似,确实为CD罕见病例。
针对该例患者鉴别诊断方面,其腹股沟淋巴结活检及骨髓活检病理均提示大量浆细胞浸润,且腹股沟淋巴结组织免疫组化提示IgG4(+)/IgG(+)为30%,因此与IgG4-RD相鉴别是非常重要的,因为两种疾病的治疗方法是完全不同的。
IgG4-RD有其自身特点,实验室检查方面:通常血清 IgG4 水平会有显著增高(>1350mg/L),外周血嗜酸粒细胞计数、比例及 IgE水平多有明显升高,而IL- 6 水平及 CRP 水平处于正常范围内或仅有轻度升高。
IgG4-RD病理特征包括以下3个方面:
(1)大量淋巴浆细胞浸润;IgG4-RD 相关疾病病变器官组织的免疫组化染色会发现 IgG4( + )淋巴浆细胞浸润,如果每高倍视野可见超过30个IgG4( + ) 浆细胞,IgG4阳性/IgG阳性细胞比例>40% 对诊断有高度的提示作用,其诊断的敏感性和特异性均显著高于血清 IgG4 水平增高。
(2)纤维化,特征性的形态为席纹状;
(3)闭塞性静脉炎 [21, 22]。
影像学方面:部分IgG4-RD患者的肺组织病理学类型为机化性肺炎或NSIP(非特异性间质性肺炎)。
我们认为
1、此例患者IL-6、ESR、CRP明显升高,血清及肺泡灌洗液嗜酸性粒细胞无升高,且淋巴结、肺组织病理未发现席纹状纤维化及闭塞性静脉炎,故支持IgG4-RD依据不充分。
2、IgG4-RD是一种自身免疫介导的炎性纤维化疾病,常见的临床症状包括硬化性涎腺炎、肺部炎性假瘤和间质性肺炎、自身免疫性胰腺炎、纤维性纵膈炎、腹膜后纤维化、硬化性胆管炎、硬化性胆囊炎等,本例患者并无上述相关系统表现,故可进一步排除此病。
3、IgG4-RD通常应用糖皮质激素治疗是有效的 [23],但本例患者应用醋酸泼尼松片治疗1月后综合评估病情并无缓解,且双肺结节及双肺间质纤维化进展,故进一步不支持IgG4-RD的诊断。
结语
CD是一种慢性淋巴组织增生性疾病,其在发病机制、临床及影像学方面表现复杂且具有非特异性。当患者表现为肺内单发较大的软组织肿块以及纵膈淋巴结肿大,临床鉴别诊断时会经常考虑到CD,但对于出现肺实质累及时仍需警惕MCD的可能,这就要求我们临床医师突破固性思维,同多学科协作,争取做到早诊断、早治疗。
参考文献
[1] CASTLEMAN B, TOWNE VW. Case records of the Massachusetts General Hospital: Case No. 40231. N Engl J Med. 1954. 250(23): 1001-5.
[2] An J, Lichtenstein AK, Brent G, Rettig MB. The Kaposi sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element. Blood. 2002. 99(2): 649-54.
[3] Keller SA, Hernandez-Hopkins D, Vider J, et al. NF-kappaB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo. Blood. 2006. 107(8): 3295-302.
[4] Polizzotto MN, Uldrick TS, Wang V, et al. Human and viral interleukin-6 and other cytokines in Kaposi sarcoma herpesvirus-associated multicentric Castleman disease. Blood. 2013. 122(26): 4189-98.
[5] Herrada J, Cabanillas F, Rice L, Manning J, Pugh W. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med. 1998. 128(8): 657-62.
[6] Bélec L, Mohamed AS, Authier FJ, et al. Human herpesvirus 8 infection in patients with POEMS syndrome-associated multicentric Castleman's disease. Blood. 1999. 93(11): 3643-53.
[7] Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972. 29(3): 670-83.
[8] Soumerai JD, Sohani AR, Abramson JS. Diagnosis and management of Castleman disease. Cancer Control. 2014. 21(4): 266-78.
[9] Bélec L, Authier FJ, Mohamed AS, Soubrier M, Gherardi RK. Antibodies to human herpesvirus 8 in POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, skin changes) syndrome with multicentric Castleman's disease. Clin Infect Dis. 1999. 28(3): 678-9.
[10] Fazakas A, Csire M, Berencsi G, et al. Multicentric plasmocytic Castleman's disease with polyneuropathy, organomegaly, endocrinopathy, M protein, skin changes syndrome and coexistent human herpes virus-6 infection--a possible relationship. Leuk Lymphoma. 2009. 50(10): 1661-5.
[11] van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018. 132(20): 2115-2124.
[12] Zhang X, Rao H, Xu X, et al. Clinical characteristics and outcomes of Castleman disease: A multicenter study of 185 Chinese patients. Cancer Sci. 2018. 109(1): 199-206.
[13] NCCN Clinical Practice Guidelines in Oncology.Non-Hodgkin Lymphoma 2015 .
[14] 王仁贵, 王仪生, 唐光健等. 胸部Castleman病的X线和CT表现与病理对照. 临床放射学杂志. 2002. 21(8): 605-608.
[15] 顾峰, 赵秀平, 邵成伟. Castleman病的影像学诊断. 肿瘤影像学. 2014. (4): 313-315.
[16] Kligerman SJ, Auerbach A, Franks TJ, Galvin JR. Castleman Disease of the Thorax: Clinical, Radiologic, and Pathologic Correlation: From the Radiologic Pathology Archives. Radiographics. 2016. 36(5): 1309-32.
[17] Johkoh T, Müller NL, Ichikado K, et al. Intrathoracic multicentric Castleman disease: CT findings in 12 patients. Radiology. 1998. 209(2): 477-81.
[18] McAdams HP, Rosado-de-Christenson M, Fishback NF, Templeton PA. Castleman disease of the thorax: radiologic features with clinical and histopathologic correlation. Radiology. 1998. 209(1): 221-8.
[19] 田欣伦, 葛莉, 冯瑞娥等. 累及肺实质的多中心性巨淋巴结增生的临床及病理特征. 中华结核和呼吸杂志. 2014. 37(5): 337-342.
[20] 赵爽, 万影, 黄子星, 余建群, 张文燕. 多中心型Castleman病的CT表现特征及其病理学基础. 放射学实践. 2018. 33(03): 299-303.
[21] Smyrk TC. Pathological features of IgG4-related sclerosing disease. Curr Opin Rheumatol. 2011. 23(1): 74-9.
[22] Ryu JH, Sekiguchi H, Yi ES. Pulmonary manifestations of immunoglobulin G4-related sclerosing disease. Eur Respir J. 2012. 39(1): 180-6.
[23] Sasaki T, Akiyama M, Kaneko Y, et al. Distinct features distinguishing IgG4-related disease from multicentric Castleman's disease. RMD Open. 2017. 3(1): e000432.
作者介绍

陈娟
(通讯作者)医学博士,主任医师,副教授,硕士生导师,宁夏医科大学呼吸与危重症医学科科室副主任,中华医学会呼吸分会第十届青年委员、第一届中华预防医学会呼吸专委会委员等。

张科东
(通讯作者)医学博士,副主任医师,副教授,西部之光学者,硕士研究生导师,中国残疾人康复协会肺康复专业委员会委员,宁夏职业病专业委员会常委委员,宁夏肺血管专业委员会委员,《中华全科医学》杂志编委。
郭晓桐
(第一作者)医学硕士,主治医师,宁夏医科大学呼吸与危重症医学科,宁夏医学会健康管理学分会秘书,宁夏医学会呼吸病学分会会员。

苏雪媛
(第一作者)医学硕士,住院医师,宁夏医科大学。